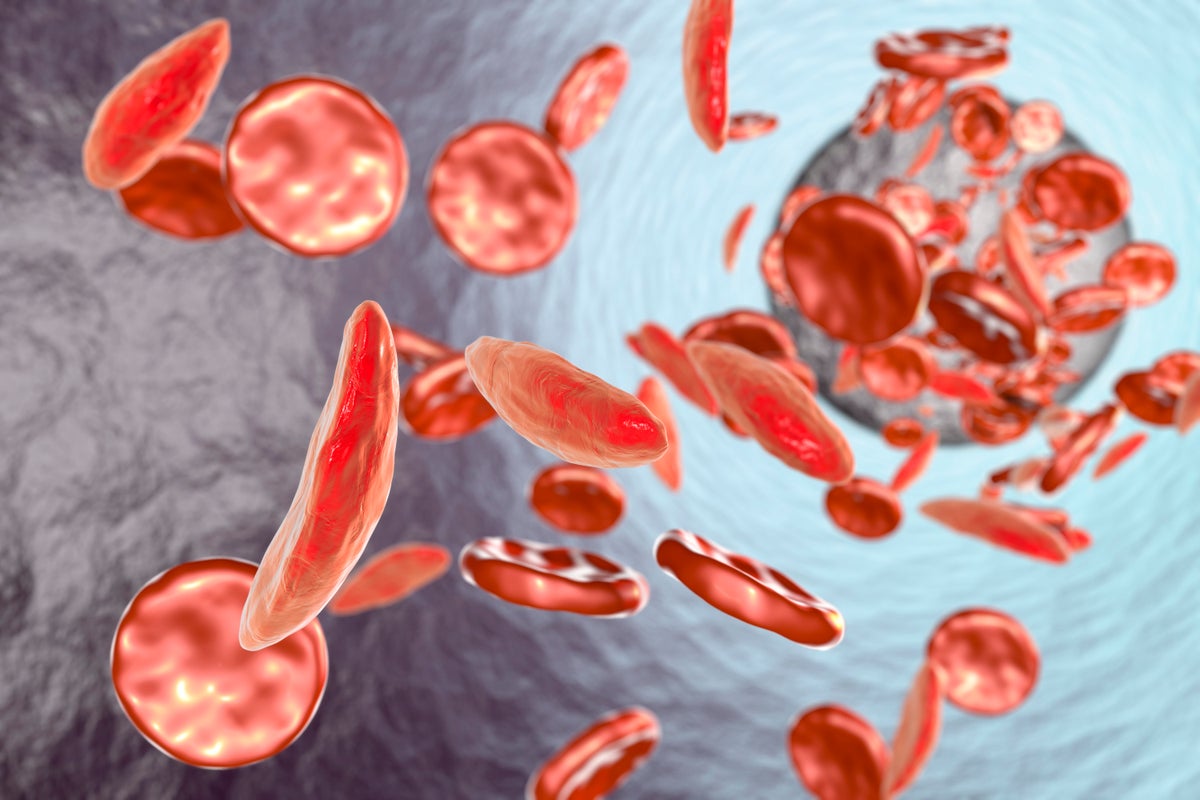
Sickle cell disease is the scourge a person’s red blood cells. The inherited blood disorder, which disproportionately affects people in sub-Saharan Africa and India, can cause unbearable pain “crises” and extreme exhaustion. And until recently, there was no curative treatment. Now approved gene therapies for sickle cell disease (including sickle cell anemia, the most extreme form) and its milder cousin, beta-thalassemia, show enormous promise.
The therapies work by deactivating or replacing a hemoglobin gene so that a person’s body makes a healthy form instead of the telltale sickle-shaped red blood cells that define sickle cell disease or averting the red blood cell deficiency that causes beta-thalassemia.
At some point, all humans produce two forms of hemoglobin, the red blood cell protein that binds oxygen so it can be transported throughout the body: a fetal form, which is more efficient at extracting oxygen in the womb, and an adult form. After we’re born, our body switches from producing the fetal form to making the adult form.
On supporting science journalism
If you’re enjoying this article, consider supporting our award-winning journalism by subscribing. By purchasing a subscription you are helping to ensure the future of impactful stories about the discoveries and ideas shaping our world today.
After years of research, scientists figured out that by turning off BCL11A—a gene known to suppress fetal hemoglobin production—they could coax the body of a person with sickle cell disease to continue making healthy hemoglobin. Companies have now developed gene therapies that target this gene. In clinical trials, people who received the treatment were functionally cured of their condition—those with sickle cell disease saw a complete resolution of their pain during the study period, and those with beta-thalassemia didn’t need blood transfusions or bone marrow transplants.
On April 18 a Breakthrough Prize in Life Sciences—one of the $3-million Breakthrough Prizes, sometimes referred to as the “Oscars of science”—was awarded to Swee Lay Thein and Stuart Orkin, who led efforts to identify the BCL11A gene and to show that shutting it off could restore healthy hemoglobin production, setting the stage for treating these devastating blood diseases.
Scientific American spoke separately with Orkin, a professor of pediatrics at Harvard Medical School and an investigator at the Dana-Farber Cancer Institute and Boston Children’s Hospital, and Thein, a senior investigator at the National Institutes of Health about what happened in the work that led to their prize and how these treatments can be made more accessible to the people who stand to benefit the most.
[An edited transcript of the interviews follows.]
How did you come to study sickle cell disease? And did you realize early on that fetal hemoglobin would be a good therapy target?
ORKIN: I started out in the 1980s working on the genetics of [beta-thalassemia]—that is, what mutations lead to the deficiency of hemoglobin in that disorder. The hope was that we would learn how a red [blood] cell is made and how genes are regulated. We didn’t really learn that, but we learned a lot about mutations and disease. Even prior to that, we knew the deficiency of beta-globin [a component of the adult hemoglobin protein] in [beta-thalassemia] and the [effects of a] mutation in sickle cell disease can be alleviated by expressing more fetal hemoglobin.
We knew that, from family studies in some very rare individuals who had a lot of fetal hemoglobin, if you raise the level of fetal hemoglobin high enough, you can basically ameliorate those disorders—plus, fetal hemoglobin is perfectly fine to substitute for adult hemoglobin [for carrying oxygen]. As early as genes were cloned back in the early 1980s, one of the goals was to see if we could reverse the switch and make fetal hemoglobin expressed at a high level in adult cells as a treatment [for beta-thalassemia]. The problem was, we didn’t understand the process at all—that’s what’s consumed the past 15 to 20 years or research—or how to reverse it.
Why do our cells switch from making fetal to adult hemoglobin in the first place?
ORKIN: We do that because, in utero, having a fetal hemoglobin is better at extracting oxygen from the mother’s circulation, and it has a higher affinity, so it takes oxygen from the circulation to the developing [fetus]. But it turns out the difference between fetal hemoglobin and adult hemoglobin in that affinity is relatively small, so having fetal hemoglobin as an adult doesn’t matter. If you ran a marathon on the top of Mount Everest, it might make a difference. You might have trouble releasing the oxygen, but under normal circumstances, it’s not a problem.
How did your work lead to the discovery of the BCL11A gene involved in sickle cell disease and beta-thalassemia?
THEIN: The discovery of BCL11A was the result of more than two decades of work driven by a deceptively simple clinical observation: Why do some people with beta-thalassemia have remarkably mild disease, while the majority require lifelong blood transfusions?
I began collecting blood samples from people with unusually mild beta-thalassemia (thalassemia intermedia) and their families. And sure enough, it turned out that most of these milder cases possessed an innate ability to produce high levels of [fetal hemoglobin]. Crucially, our family studies showed that the responsible gene or genes were inherited independently of the beta-globin gene itself and that the inheritance pattern was complex. I was convinced that that there was a substantial genetic component underlying this common [fetal hemoglobin] variation, which I confirmed with twin studies.
Then genome-wide association studies revealed the involvement of BCL11A, a gene with no previously known role in hemoglobin biology. Our findings were independently confirmed by another group the following year, firmly establishing BCL11A as a key regulator of fetal hemoglobin and, ultimately, a therapeutic target in both sickle cell disease and beta-thalassemia.
ORKIN:Back in 2011 we did an experiment in which we took mice that had been engineered to have sickle cell anemia and disabled the BCL11A gene in those mice—but only in the developing red blood cells—through fancy genetics. The result was that we could completely correct those mice—they were completely well after we knocked out [deactivated] the BCL11A gene. That told us that one gene was sufficient to correct the disease and that it would be a therapeutic target if we could manipulate it. That was 15 years ago. It took several years to figure out where we’d want to do the editing, and just about the time we wanted to ask that question, [the gene-editing technique] CRISPR came on the scene, so, you know, all the stars aligned in just the right way.
Dr. Thein, can you describe some of your research with populations in Malawi?
THEIN:At the time, identifying the genes responsible for elevated fetal hemoglobin relied on a technique that requires large, multigenerational family cohorts with well-documented relationships. Finding such families is no small feat, so when I came across a person with exceptionally mild beta-thalassemia who happened to come from a remarkably large extended family, many of [whose members] were living in Malawi, I recognized it as a rare and significant opportunity.
I organized a field trip to Malawi and, through careful tracing and recruitment, was able to expand the study family to 210 individuals spanning seven generations—an extraordinary resource for this technique.
There was some hesitation among family members initially, which is entirely understandable when people are asked to participate in something unfamiliar. We addressed this through clear, patient explanations of the study’s purpose and what participation involved.
What had begun as a scientific endeavor became, in many ways, a communal one—a reminder that behind every dataset are real people whose generosity and trust make the research possible.
These discoveries paved the way for the first approved gene-editing treatments for sickle cell disease in 2023: Casgevy, made by Vertex Pharmaceuticals and CRISPR Therapeutics, and Lyfgenia, made by bluebird bio (now known as Genetix Biotherapeutics). How many people with sickle cell disease have received these therapies?
ORKIN:The original [Vertex] trial had [about] 75 participants [with either sickle cell disease or beta-thalassemia, and since then, they’ve treated more people. They report that more than 90 percent of the participants who were treated are basically functionally well. In other words, in the case of [beta-thalassemia], they don’t need transfusions anymore, and in terms of sickle cell disease, they don’t get sickle crises, the painful crises.
It really is transformative for these individuals, particularly for the people with sickle cell. Beforehand, it was a miserable disease. They had intermittent pain crises and other complications. And it’s hard to maintain a job if you’re an adult. And what the patients describe is, after they’re treated, they have a new lease on life.
Are the populations most at risk for these diseases likely to receive gene therapy treatment for their conditions? And how can these treatments be made more affordable and accessible?
THEIN:Honestly, in the near term, the answer is probably not.
The gene therapies currently approved by the U.S. Food and Drug Administration are ex vivo. This means harvesting a patient’s own hematopoietic [red blood] stem cells, editing them in a specialized laboratory and then reinfusing them—but only after the patient has undergone intensive chemotherapy to destroy the existing bone marrow and create space for the edited cells to engraft [settle and begin producing new cells]. The process is physically grueling for the patient, logistically demanding and extraordinarily expensive, costing about $2 million to $3 million per patient. Even in the wealthiest health care systems, access is far from universal.
The scientific community is acutely aware of this, and research priorities are now pivoting toward next-generation in vivo gene-editing approaches where the editing machinery is delivered directly into the body to target the hematopoietic stem cells in situ.
But the challenge that weighs on me most is access to treatment, whether [it is] gene therapy, [a] bone marrow transplant or drugs. The burden of sickle cell disease is heaviest in sub-Saharan Africa and India, precisely where these therapies are currently least accessible. Even if we develop a cheaper, simpler gene therapy tomorrow, getting it to the patients who need it most will still require political will, sustained global health investment, international partnerships and a serious rethinking of how we price and distribute transformative medicines.
What are you working on next?
ORKIN:My group is focused on trying to understand in very exquisite detail the whole mechanism and the process that is involved in the switch [from fetal to adult hemoglobin]. And we’re focused on trying to see if we can develop a way to find small molecules that will do the reversion, if you will, by taking a pill. That would be something that could be distributed much more easily than the current editing therapy.
THEIN: My current research is centered on small molecules, particularly those that can prevent or abort the severe pain crises that remain one of the most debilitating and undertreated aspects of sickle cell disease. These crises represent a profound unmet clinical need, and finding effective, accessible interventions for them would make an enormous difference to patients’ daily lives.